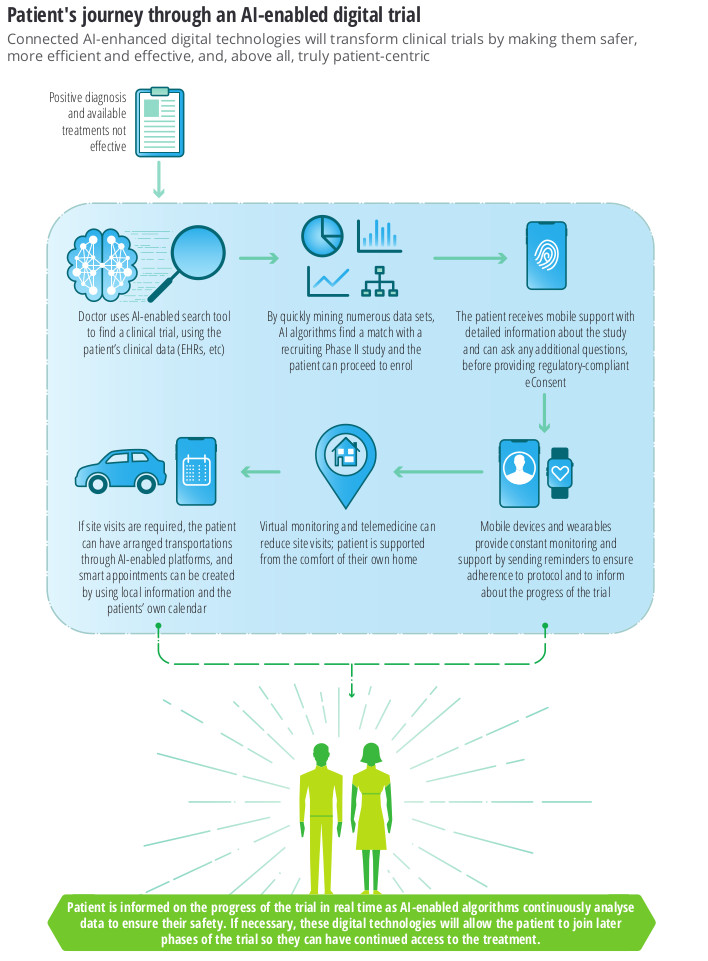
Why is it so hard to automate the analysis of a regulated clinical trial?
Sure, you might be able to setup an EDC in the blink of an eye, but lets be honest – it still takes a team of people years of effort to analyse the results and produce the tables, figures and listings (TFL) that go into the Clinical Study Report.
You’ve done a proof-of-concept or two, maybe piloted automation of SDTM datasets, and have bought into the end-to-end automation vision.. but how do you get there from here? And why does it seem so hard?
Automation is the tip of the iceberg
The first thing to recognize is that automation is not standalone. To achieve end-to-end automation in a production environment means that all the necessary metadata is available at each step of the process; the key people have the tools and skills to curate and manage that metadata; and they need a robust and validated software platform that meets regulatory submission and GCP quality requirements.
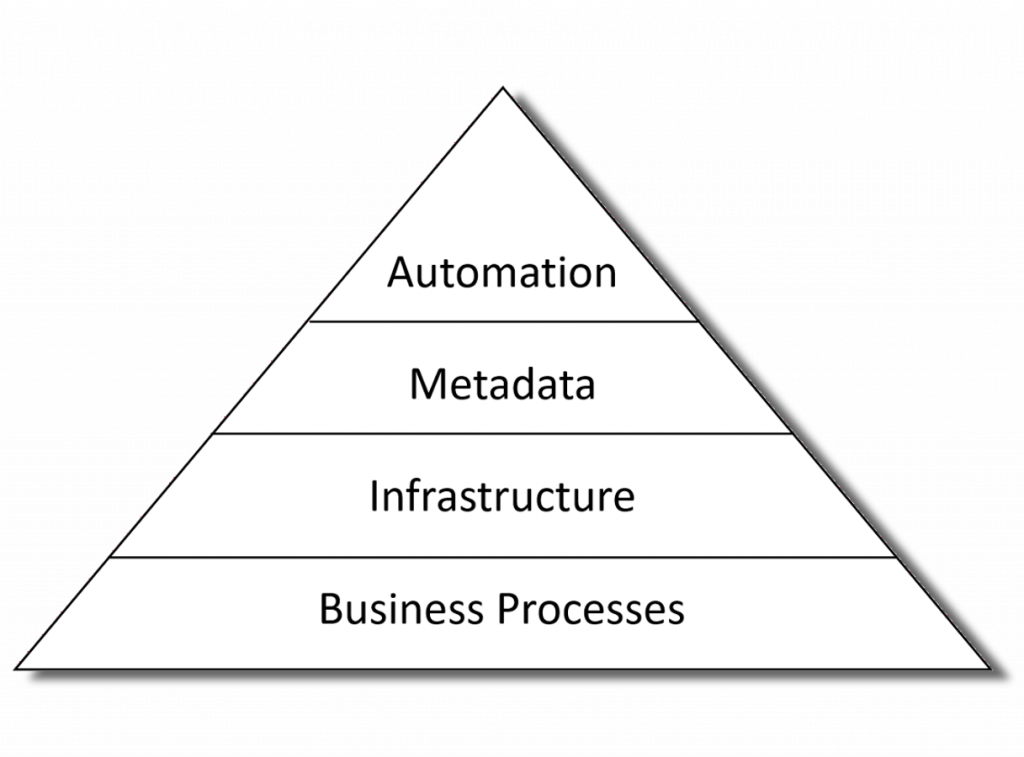
Ok, so lets looks as each of the areas than are needed to support successful automation of analysis results.
Don’t build a horse-less carriage
Before Henry Ford started building the Model T using a production line, cars were build in the same was as horse-drawn carriages has been – by a small team of highly skilled people that worked on the product from beginning to end.
Sound familiar?
Change and disruption caused by the introduction of automation is well understood in manufacturing, however it is a new phenomenon in knowledge-based industries.
Don’t re-invent the wheel either
The software industry pioneered Model-Driven Development (MDD) twenty years ago, and there now exist ISO Standards for Model-Driven Architecture, methodologies and tools that have been used successfully in regulated, safety-critical industries such as avionics, space, energy, etc.
Adoption of Model Driven Development can be mapped out using the Capability Maturity Model (see: MDD Maturity Model)
| Level | Maturity | Description |
| 1 | Ad-Hoc | Analyses are not model-driven |
| 2 | Basic | Basic use of models in organization. |
| 3 | Initial | The organization starts developing systems more according to model model-driven approach. |
| 4 | Integrated | Models at different abstraction levels are built and they are fully integrated within a comprehensive modelling framework |
| 5 | Ultimate | The role of coding will disappear and the transformations between models are automated. |
So, while most organizations will be starting at level 1 (i.e. Analyses are created manually), the question is: What level of adoptions do you aspire to? and over what timescale?
Then you can plan how to transition to level 2 – i.e. basic metadata-driven automation running end-to-end through the analysis.
Model-driven architecture
Possibly the clearest way of describing the architecture required to support a fully automated analysis using a Four layered metamodel architecture:
| Layer | Contents | Description |
| M3 | Meta-metamodel | Metadata standards, e.g. W3C Semantic Web, XML, UML, ISO11179, etc. |
| M2 | Metamodel | Clinical Standards, e.g. CDISC, MEdDRA, SNOMED, etc. |
| M1 | Model | Trial-specific Metadata |
| M0 | Data | Clinical, Rando, Labs Data, etc. |
Each level of the Metadata Architecture requires its own set of tools and processes, and people with the skillset to be able to work at the relevant level of abstraction.
Taking the first step
While a full-blown end-to-end level-5 CMM Ultimate Meta-Model Architecture may remain a powerpoint vision, there is more than enough work involved in getting from level-1 to level-2.
A full list is clearly out of the scope of this blog post(!), however, some things to consider include:
- Statisticians will need to be able to create a SAP in a machine-readable format. What tools will they use? who will train and support them?
- Programmers will need to be able to work with technologies such as XML, RDF,/OWL, Relational and Graph databases.. and probably using languages other than SAS
- How is study-specific data handled? How do you work around partial and dirty data?
- Software infrastructure to support version control, continuous integration/deployment, metadata repositories
- How will validation be done without ‘mainline’ and ‘qc’ programs?
- Business processes will be needed to support a software development and DevOps
- How will Industry Standards (e.g. CDISC) be managed? What about Corporate and Study/Drug/Disease-Specific standards? What happens when a new version of a standard is issued?
None of these issues are insurmountable, but equally they add up to more than an quick fix.
Summary
The promise of end-to-end automated analysis of clinical trials is not just that it will be faster and easier, but also the new and innovative applications that will be unlocked (CMM Level 3 and beyond).
Key points to consider in planning an automation strategy are:
- Automation requires metadata, and that means new tools, skills and business processes.
- This is mostly a solved problem, but will require learning lessons from other industries
- While implementing end-to-end automation requires work, ultimately is means that you spend less time on grunt-work and more time on activities that add value and ultimately help patients!