A great question which speaks to real concerns and forces us to examine why we are automating in the first place, and what we hope to achieve.
Increasing employment rates
The World Economic Forum estimates that by 2025, technology will create at least 12 million more jobs than it destroys, a sign that in the long run, automation will be a net positive for society
By “augmenting human intellect” we mean… more rapid comprehension, better comprehension, the possibility of gaining useful degree of comprehension in situation that previously was too complex, speedier solutions, better solutions, and the possibility of finding solutions to problems that before seemed insoluble.
Introduction to Augmented Human Intellect: A Conceptual Framework
The goal is to optimize comprehension, not to reduce or eliminate work.
Automation is one of the tools that we use to improve comprehension – it allows us to crunch more data, more quickly, and to visualise it more easily.. all of which helps reduce “time to insight”.
Automation transforms jobs
So, to address the original question – will automation replace your job? No. However is will certainly change your job.
The tools and processes that we use to design clinical trails, specify what data to collect, how to analyse the data and report on trials will all change.
The goal (of automation) is not (to employ) less people; The goal is better comprehension, more quickly.
The Tower of Babel by Pieter Bruegel the Elder. ARS ELECTRONICA Deep Space 8K
Behold, the people is one, and they have all one language… and now nothing will be restrained from them, which they have imagined to do.
Genesis 11:6
A compelling vision
Clinical Trial analysis and reporting is changing, driven by a vision of end-to-end automation which aims to substantially improve efficiency, consistency, and re-usability across the clinical research data lifecycle.
Conferences are a buzz with debate on the best languages to use to implement this future vision – will SAS prevail? or open-source languages like R or Python?..
Addressing the challenge of automation by talking about programming languages is like looking at the Moon with a microscope!
Programmer Proverb
This article aims to look at the heavens using a telescope!.. OK, enough with the analogies!
Let’s start by trying to understand what exactly the challenge is with automation?
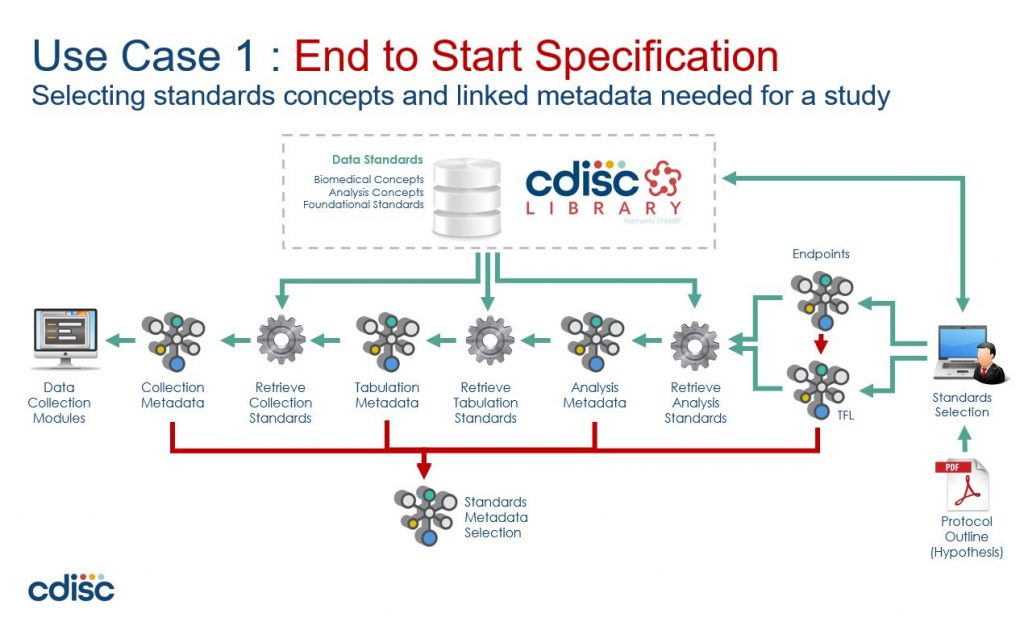
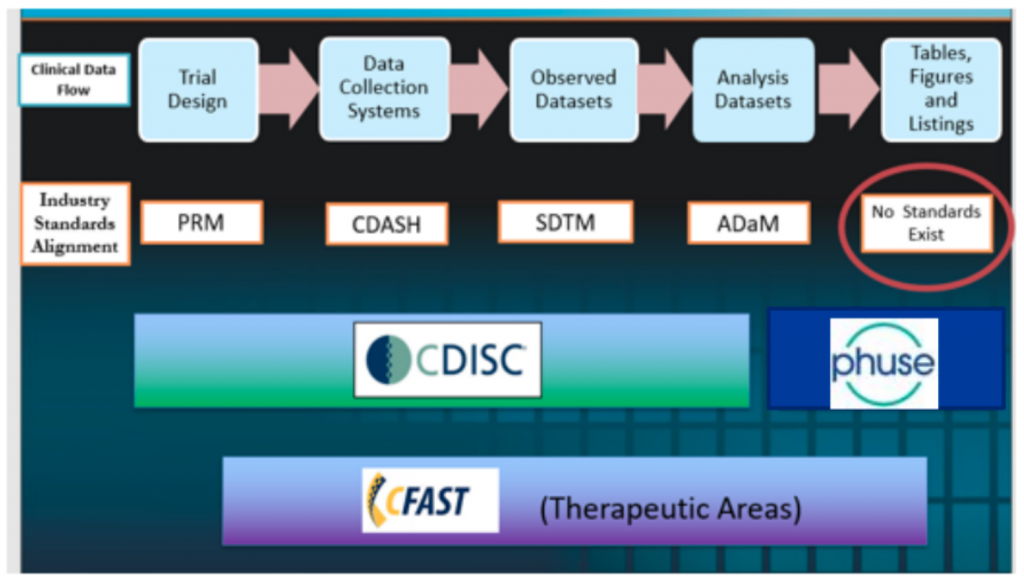
CDISC 360 Vision of end-to-end automation
Trouble in paradise
Lets consider a typical automation project:
First, you build a proof-of-concept – maybe an R-Shiny front-end onto a SAS backend that re-uses macros that you’ve been using for years. Everything looks good, ‘click-a-button’ demos, great powerpoints, and everyone is excited… so onto the next stage…
Next is a pilot project – you need something more robust (did someone mention validation??).. Build a web interface in Python, put the metadata in a (graph?) database with the remnants of the PoC (which has now been tested) driving the SAS backend.
Perfect! Now you can see the benefits of “substantially improved efficiency, consistency, and re-usability”.. Can’t you?
What do you mean no? Why not?
Well, because there still seems to be a lot of manual work managing all that metadata.
The problem is that while we no longer have to manually create and configure a suite of programs, now instead, we have to manually create and configure all the metadata that drives the automation.
So, the challenge of automation is actually the challenge of how to efficiently manage metadata.
The tower of Babel
So, lets now examine the source of this inefficiency. We can trace it back to a couple of key assumptions:
A clinical trial involves many specialist roles – Investigators, Clinicians, Regulators, Data Managers, Statisticians, etc. Each with their own definitions and terminology – their own domain-language. The assumption that a single common data model can fit all these specialist sub-domains results in a data model that is unwieldy, hard to use and even harder to change.
The assumption that metadata must be stored in a central metadata repository (MDR) leads to a ‘database-centric architecture’ which is problematic in its own right, and also has increased cost of ownership (e.g. managing and maintaining the database server) – not to mention the ubiquitous form-based user interface that needs an endless series of click-and-wait manual steps for every task
These assumptions lead to a ‘tower of babel’ scenario where we have a common data model that is stretched in every direction trying to support every nuance of every domain-expert, and is stored in a central database which becomes increasingly inefficient to use and manage as it gets wrapped in IT processes and change requests.
What we need is an ‘de-centralised’ approach that does not have a single common model in a central repository
Confound their language
To explore what such a ‘de-centralised’ approach might look like, let’s walk through an example using the BRIDG model
The goal of the BRIDG Project is to produce a shared view of the semantics of a common domain-of-interest, specifically the domain of basic, pre-clinical, clinical, and translational research and its associated regulatory artefacts.
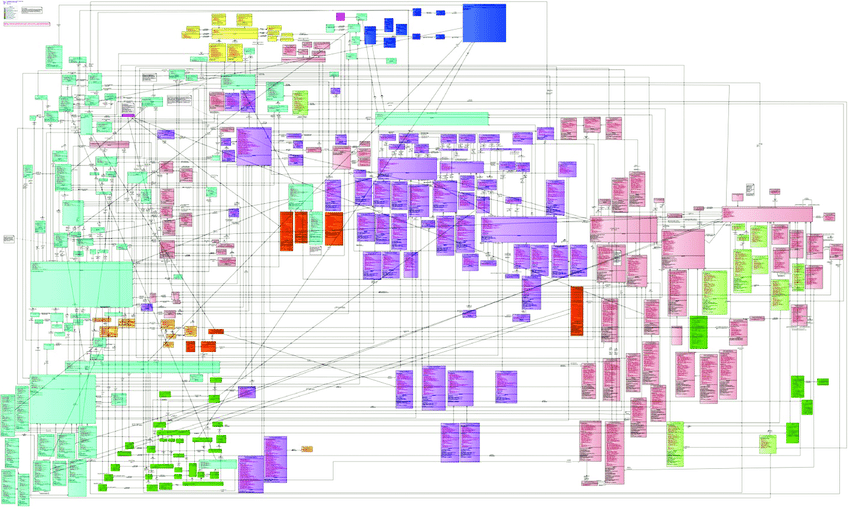
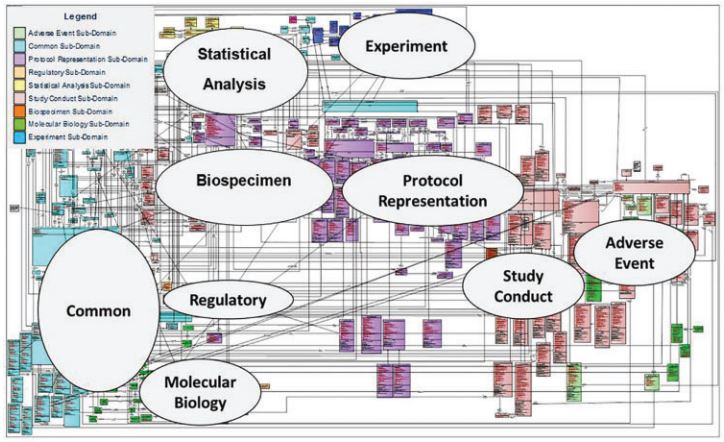
The diagram below shows the BRIDG model in its full glory. If this one diagram does not communicate the complexity of a single domain model then nothing will!
To be fair, BRIDG make it clear that this comprehensive view is published to check model integrity, and users are recommended to use the sub-domain views to understand the model.
The fact that BRIDG recommends sub-domain views confirms the complexity of a common model, but also identifies a solution – simply split the common model into parts!
The annotations in the diagram above are simply drawn onto the image – they are not part of the model, do not describe the relationship between sub-domains, and are not machine readable.
The BRIDG model is written in Unified Modelling Language (UML), a general-purpose modelling language intended to provide a standard way to visualize the design of a software system.
What we need is a “meta” language that sits “above” UML, to describe component parts and relationships between these parts.
Positive space
More specifically, we want to use this “meta language” to:
Model different sub-domains of the clinical trials lifecycle that have specific terminology and definitions used by domain expert working within that sub-domain, because e.g. Statistician’s speak a different language than Investigators, or Data Managers, etc.
De-couple sub-domains and describe relationships between them and how they are shared and translated. For example both the Experimental and the Statistical Analysis sub-domains have the concept of an unscheduled visit, but each treats them differently, however we still want to link and translate between the two.
Great – so now we know what we want to do with our “meta language”.. but what is it?…
illustration of Christopher Alexander’s ‘Positive Outdoor Space’ by Matt Noiseux
A pattern language
A pattern language is an organized and coherent set of patterns, each of which describes a problem and the core of a solution that can be used in many ways within a specific field of expertise.
The term “pattern language” was coined by architect Christopher Alexander in his 1977 book A Pattern Language.
Although the original concept came from building architecture and urban planning, pattern languages have since been used in many other disciplines – and since the 1990s for software design patterns.
One such software pattern language is Domain-Driven Design (DDD), a term coined by Eric Evans in 2003 in his book of the same name.
Domain-Driven Design
Domain-driven Design is a software design approach focusing on modelling software to match a domain according to input from that domain’s experts.
Well, that sounds promising! Let’s see if we can apply Domain-Driven Design to the BRIDG model…
DDD uses context maps to describe how a domain is divided into parts and the relationships between those parts.
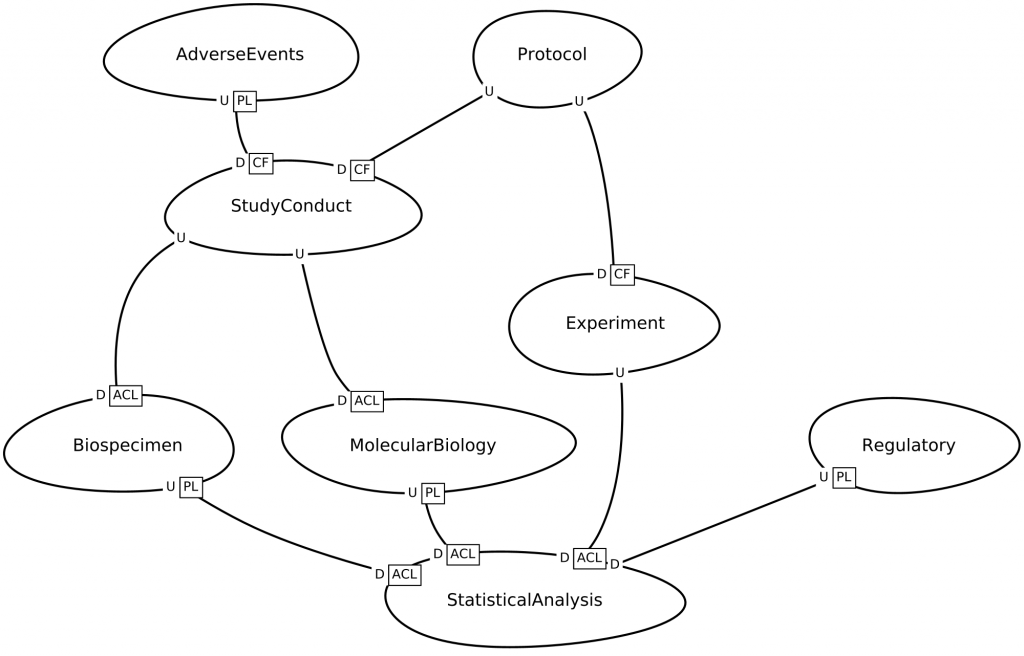
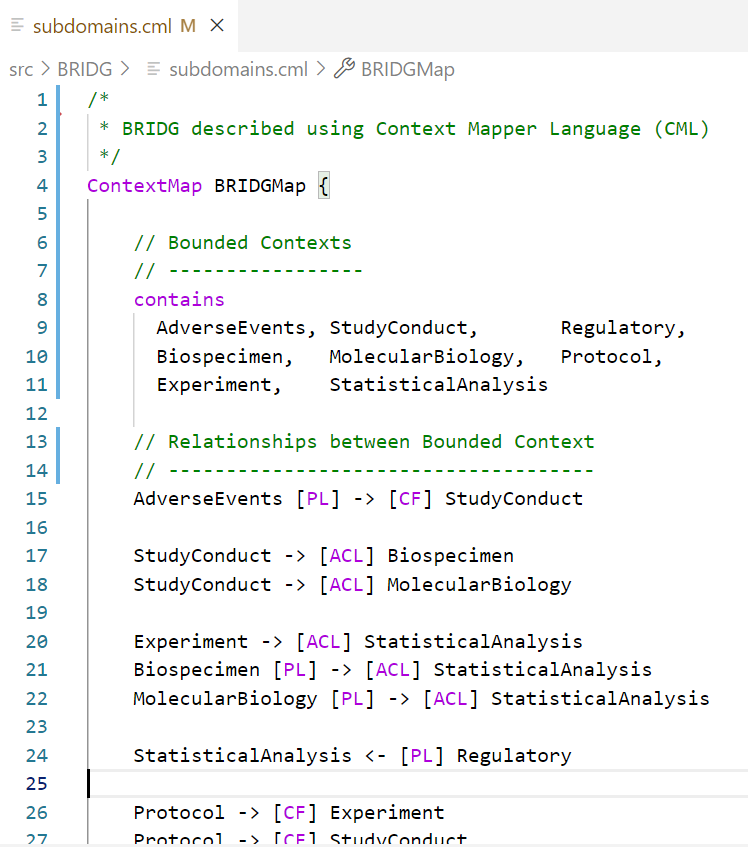
Consider the BRIDG context map below, which was generated from a description written in the Context Mapper language. Notice how this resembles the informal annotations on the UML diagram above.
BRIDG context map, bounded contexts and relationships
However, this context map is fundamentally different from the annotated UML diagram above, because:
This context map is generated using a domain-specific language (DSL), which means that the pattern language is an actual ‘computer language’ that can be used in programming editors like VS Code or Eclipse, etc. with syntax highlighting, code-completion, etc.
Not only can the DSL be used to generate diagrams, it can also be transformed into other models – for example into microservice contracts which in turn can be used to generate code.
BRIDG context map, described using CML being edited in VS Code
So there we have it! A pattern language that allows us to pull down the Tower of Babel.
We can create models of the clinical trials domain split into component parts, and model those parts independently while managing the interfaces between them. So, we don’t need a single common data model.
We can treat metadata like program files – stored in text files that are ‘linked’ together when they are ‘run’. So, we do not need a central database to store metadata.
Metadata is machine-readable and can be transformed into different metadata files, diagrams, or program code that processes clinical data. So, we can automate end-to-end
Summary
We started off by looking for a way to deliver on the vision of end-to-end automation and achieve the benefits of substantially improve efficiency, consistency, and re-usability across the clinical research data lifecycle.
The assumption that we need a single common data model stored in a central repository leads to inefficiencies which negate the benefits of automation.
Pattern languages are commonly used in software engineering for reusable solutions to commonly occurring problems, and we walked through an example by applying Domain-Driven Design to the BRIDG model.
Using a domain-specific language (DSL), we are able to split the common data model into parts, and store each part as machine-readable metadata in text files to generate code.
This approach streamlines metadata management because we can leverage source-code processes and tools to manage metadata – syntax checking, version control, unit testing, CI/CD, etc… and not a web-form in sight!
Stay tuned for more examples and demos which are in the pipeline.. in the meantime here is some background reading:
Enabling end-to-end Automation in Analysis and Reporting
PHUSE US Single Day Event 21-MAY-2021
Summary
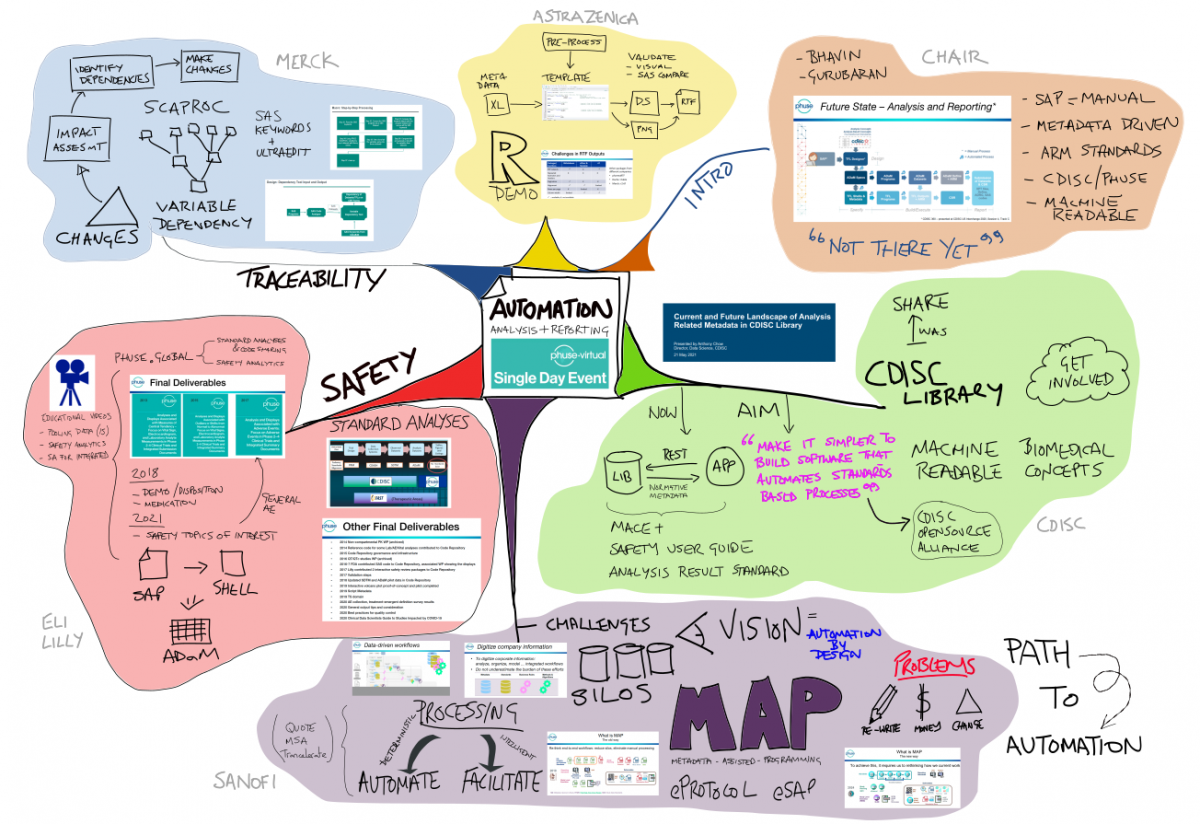
The PHUSE US Spring 2021 SDE was an interesting cross-section of perspectives on automation of Analysis and Reporting. My takeaway messages were:
End-to-end machine readable standards are still a work-in-progress, and there is not a clear path to standardisation around e-Protocol and e-SAP
CDISC aim to make it simpler to implement software that automates standards (rather than publishing standards as ‘600-page pdf documents’); this is based on the proprietary CDISC Library
Key implementation challenges include: Managing change, Budgets (it takes longer than you think) and Silo business processes
PHUSE have a mature set of safety reporting deliverables, including SAP definitions, statistical methods and visualizations. This can provide a solid basis for automation projects.
DISCLAIMER: I missed one session by Farha Feroze (Symbiance) on Automated TFL Moch Shell Generation using AI Techniques (ML/NL), so is not included in this report!
Future State
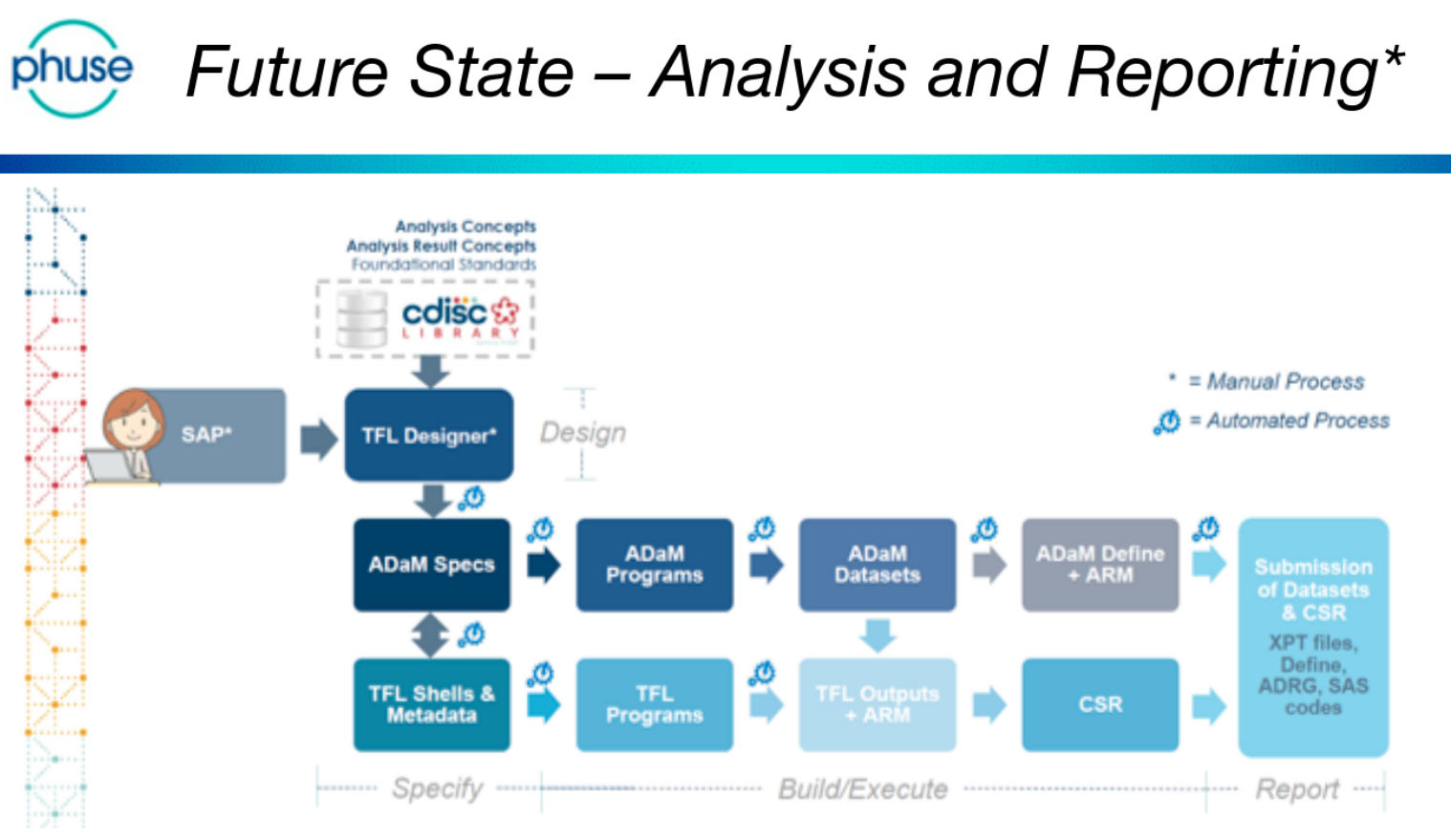
The day was (excellently!) chaired by Bhavin Busa, and Gurubaran Veeravel, who kicked-off with reference to the CDISC 360 ‘Future State – Analysis and Reporting’, noting that “we are not there yet!”
This is a future-state vision that is based on automating the current reporting pipeline. Currently ADaM and TFL programs are written manually.
CDISC Standards
Anthony Chow presented an overview of the CDISC Library, and current/future projects related to Analysis & Reporting
CDISC aim to make it simpler to build software that automates standards-based processes.
The CDISC Library provides an application programming interface (API) to ‘normative metadata’ on all CDISC standards
CDISC is working with open source projects on tools to access CDISC Library
CDISC have Analysis & Results projects ongoing and about to start recruiting members: MACE+ project, Safety User Guide, and Analysis Results (ARM) Standard development.
Path to Automation
Andre Couturier & Dante Di Tommaso (Sanofi) gave an insightful presentation on the challenges facing when implementing end-to-end automation (for a start.. don’t call it automation!)
It is key to communicate a clear vision and ‘sell upwards’
Key challenges include change management, Silo business processes, and budget (it takes longer than you think!)
Decide whether existing processes are ‘Deterministic’ or ‘Intelligent’ – Deterministic processes are candidates for automation, and Intelligent processes can be ‘facilitated’
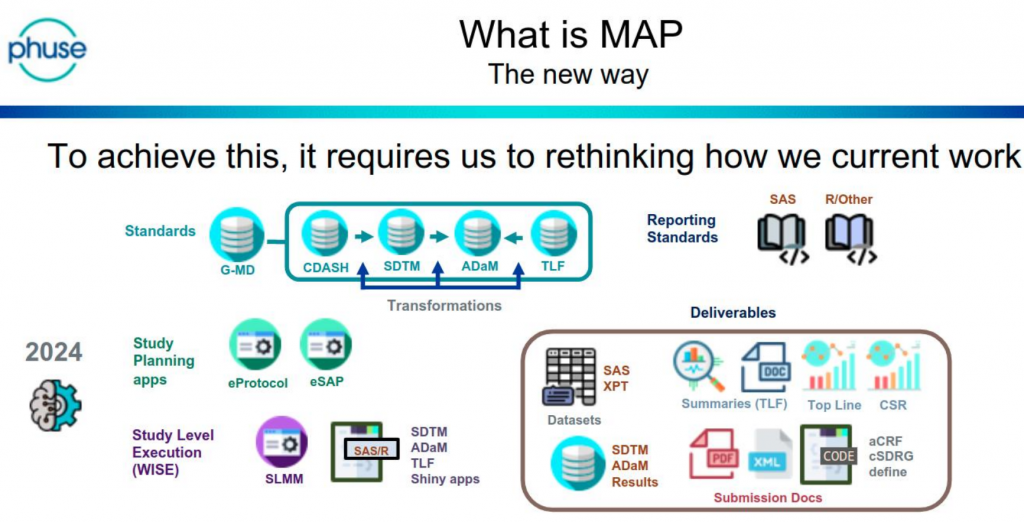
Sanofi use the acronym MAP (Metadata Assisted Programming) to describe the change in approach
Safety Reporting
Mary Nilsson (Eli Lilly) provided a comprehensive overview of the work that PHUSE has done on Safety Reporting. A comprehensive set of deliverables are available phuse.global
The two working groups are: Standard Analyses and Code Sharing (pre-2020) and also Safety Analytics (post-2020)
In addition there are training videos covering pooling data, safety analytics and for integrated reporting
The deliverables include SAP text, statistical methods, and visualisations
This is a body of knowledge that can provide a solid set of ‘source documentation’ for automation projects
Traceability and dependency
Gurubaran Veeravel walked-through how Merck perform impact analysis when standards change – specifically to determine program dependencies and variables used.
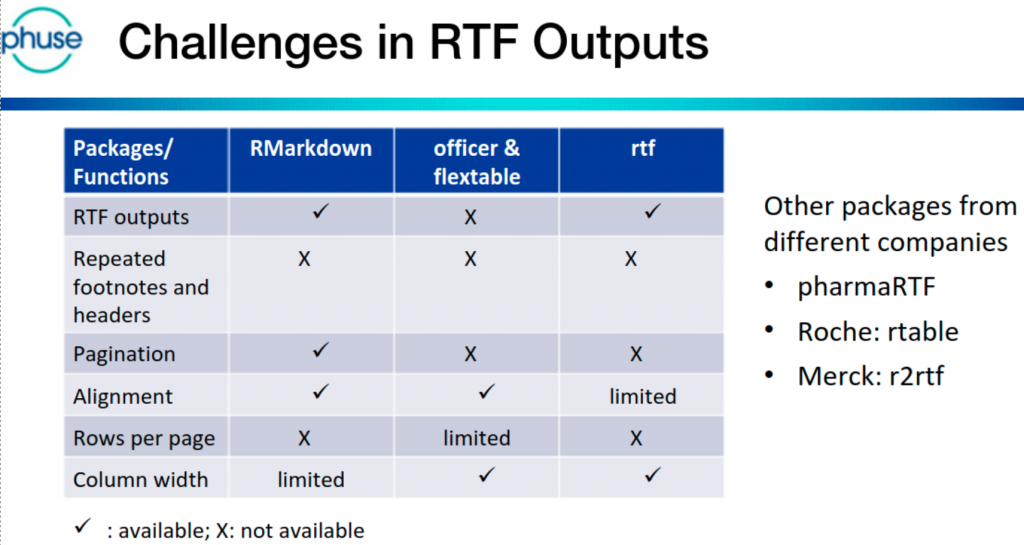
R demo – TFL Generation
Jem Chang & Vikram Karasala (AstraZeneca) presented how R programs can create RTF outputs with the same layout as existing (SAS) outputs. Alternative/existing R packages are available, and the pros/cons of each were discussed.
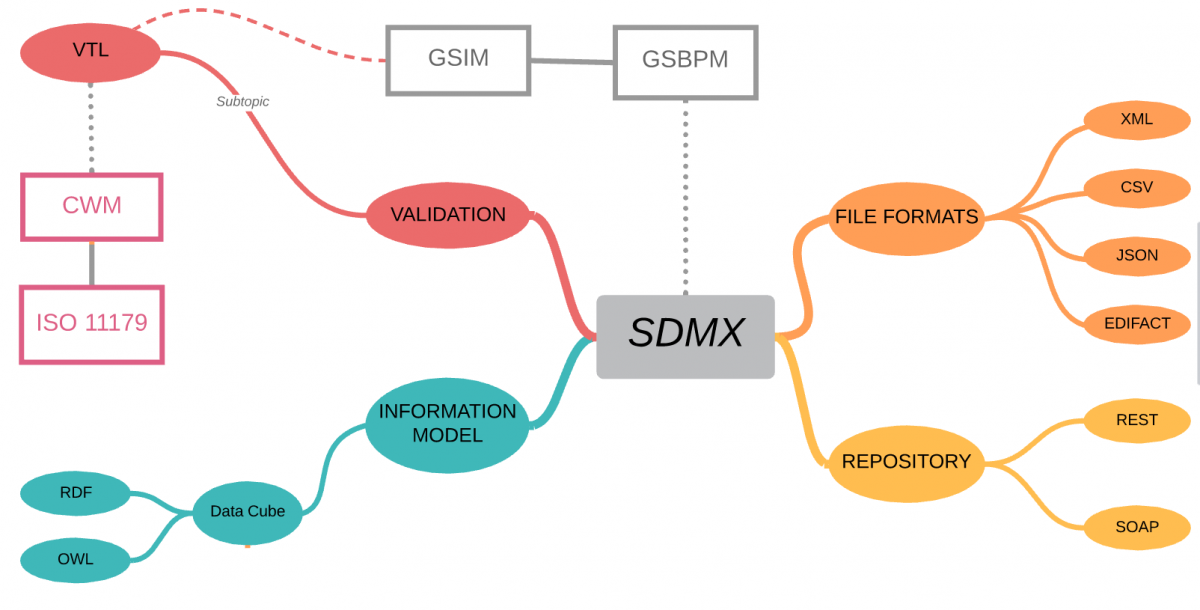
SDMX is a comprehensive, domain-neutral, ISO standard for Statistical Data and Metadata exchange, first released in 2004.
Let’s clear-up one misconception straight away – although SDMX stands for Statistical Data and Metadata eXchange – you should not let the “eXchange” part fool you into thinking that this is simply a file format – it is so much more!
The SDMX standard provides:
Technical standards (including the Information Model)
Statistical guidelines
an IT architecture and tools
Taken together, the technical standards, the statistical guidelines and the IT architecture and tools can support improved business processes for any statistical organisation as well as the harmonisation and standardisation of statistical metadata.
sdmx.org
Domain neutral
Although SDMX was established by international banking and government organisations, the information model is domain neutral, and because it is based on W3C Semantic Web standards, it aligns with the CDISC vision of linked data and biomedical concepts.
SDMX defines a vocabulary for describing Statistical data using W3C Data Cube which and so all domain-specific metadata is described using OWL ontologies – if this is new to you, then have a look at bioontology.org – The world’s most comprehensive repository of biomedical ontologies!
For this reason alone, SDMX provides a pathway to the CDISC vision of clinical trials analyses based on linked-data and biomedical concepts.
The statistical guidelines aim at providing general statistical governance as well as common (“cross-domain”) concepts and code lists, a common classification of statistical domains and a common terminology.
sdmx.org
Clinical trials typically involve data exchange across a network of Sponsors, Regulators, Vendors, Labs, CRO’s, etc. Each with different roles as data produces and consumers, different agreements on who can access what data when. These are all scenarios covered by the SDMX Statistical Guidelines and GSBPM.
Metadata repository
SDMX provides the specification for the logical registry interfaces, including subscription/notification, registration of data and metadata, submission of structural metadata, and querying, which are accessed using either REST or SOAP interfaces.
Metadata Repositories (MDR) are at early-stage adoption within Clinical Trials, so a key benefit of a standard interface is that it allows a period of experimentation/evolution on how the MDR is implemented with limited impact the rest of your analytics platform.
File interchange format
Well, yes! SDMX does also include file format standards for the exchange of data – XML, CSV, JSON are probably the key ones for use in clinical trials, allowing data transfer between languages and systems with minimal change.
Validation and Transformation Language
SDMX also includes a fully specified Validation and Transformation Language (VTL) which allows statisticians and data managers to express logical validation rules and transformations on data can be converted into specific programming languages for execution (SAS, R, Java, SQL, etc.)
Although the VTL language originated under the governance of SDMX, it was recognised that other communities could benefit and so VTL was designed to be usable in SDMX, DDI and GSIM
Summary
Why consider SDMX for use in clinical trials?
In short, because it’s a comprehensive, established standard which can be applied to any Statistical domain, and it ‘plays nicely’ with many other standards.
But what problem will it help solve? Typical use-cases might include:
Creating vizualisations for Blind Review that take place before ADaM and TFL programming is completed,
Implementation of CDISC standards using linked-data and biomedical concepts,
Improved governance of data transfers between Sponsors, Regulators, Vendors and CRO’s
Validation of open-source technologies and new languages such as R, Python or Julia,
A standards standards-based Metadata Repository (MDR) interface that can remain constant from pilot through deployment regardless of implementation technology or vendor.
Even if you do not include SDMX, there are certainly parts that are worth consideration.
Why is it so hard to automate the analysis of a regulated clinical trial?
Sure, you might be able to setup an EDC in the blink of an eye, but lets be honest – it still takes a team of people years of effort to analyse the results and produce the tables, figures and listings (TFL) that go into the Clinical Study Report.
You’ve done a proof-of-concept or two, maybe piloted automation of SDTM datasets, and have bought into the end-to-end automation vision.. but how do you get there from here? And why does it seem so hard?
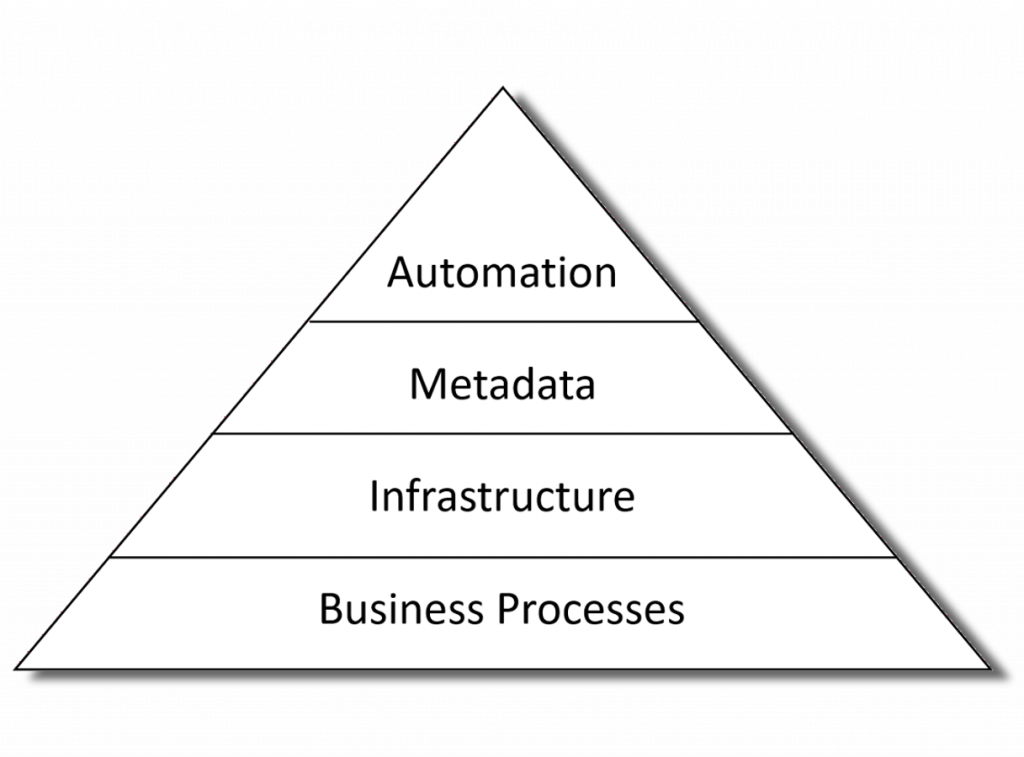
Automation is the tip of the iceberg
The first thing to recognize is that automation is not standalone. To achieve end-to-end automation in a production environment means that all the necessary metadata is available at each step of the process; the key people have the tools and skills to curate and manage that metadata; and they need a robust and validated software platform that meets regulatory submission and GCP quality requirements.
Automation requires a foundation of people, skills and tools
Ok, so lets looks as each of the areas than are needed to support successful automation of analysis results.
Don’t build a horse-less carriage
Before Henry Ford started building the Model T using a production line, cars were build in the same was as horse-drawn carriages has been – by a small team of highly skilled people that worked on the product from beginning to end.
Sound familiar?
Change and disruption caused by the introduction of automation is well understood in manufacturing, however it is a new phenomenon in knowledge-based industries.
Don’t re-invent the wheel either
The software industry pioneered Model-Driven Development (MDD) twenty years ago, and there now exist ISO Standards for Model-Driven Architecture, methodologies and tools that have been used successfully in regulated, safety-critical industries such as avionics, space, energy, etc.
Adoption of Model Driven Development can be mapped out using the Capability Maturity Model (see: MDD Maturity Model)
Level
Maturity
Description
1
Ad-Hoc
Analyses are not model-driven
2
Basic
Basic use of models in organization.
3
Initial
The organization starts developing systems more according to model model-driven approach.
4
Integrated
Models at different abstraction levels are built and they are fully integrated within a comprehensive modelling framework
5
Ultimate
The role of coding will disappear and the transformations between models are automated.
So, while most organizations will be starting at level 1 (i.e. Analyses are created manually), the question is: What level of adoptions do you aspire to? and over what timescale?
Then you can plan how to transition to level 2 – i.e. basic metadata-driven automation running end-to-end through the analysis.
Model-driven architecture
Possibly the clearest way of describing the architecture required to support a fully automated analysis using a Four layered metamodel architecture:
Layer
Contents
Description
M3
Meta-metamodel
Metadata standards, e.g. W3C Semantic Web, XML, UML, ISO11179, etc.
M2
Metamodel
Clinical Standards, e.g. CDISC, MEdDRA, SNOMED, etc.
M1
Model
Trial-specific Metadata
M0
Data
Clinical, Rando, Labs Data, etc.
Each level of the Metadata Architecture requires its own set of tools and processes, and people with the skillset to be able to work at the relevant level of abstraction.
Taking the first step
While a full-blown end-to-end level-5 CMM Ultimate Meta-Model Architecture may remain a powerpoint vision, there is more than enough work involved in getting from level-1 to level-2.
A full list is clearly out of the scope of this blog post(!), however, some things to consider include:
Statisticians will need to be able to create a SAP in a machine-readable format. What tools will they use? who will train and support them?
Programmers will need to be able to work with technologies such as XML, RDF,/OWL, Relational and Graph databases.. and probably using languages other than SAS
How is study-specific data handled? How do you work around partial and dirty data?
Software infrastructure to support version control, continuous integration/deployment, metadata repositories
How will validation be done without ‘mainline’ and ‘qc’ programs?
Business processes will be needed to support a software development and DevOps
How will Industry Standards (e.g. CDISC) be managed? What about Corporate and Study/Drug/Disease-Specific standards? What happens when a new version of a standard is issued?
None of these issues are insurmountable, but equally they add up to more than an quick fix.
Summary
The promise of end-to-end automated analysis of clinical trials is not just that it will be faster and easier, but also the new and innovative applications that will be unlocked (CMM Level 3 and beyond).
Key points to consider in planning an automation strategy are:
Automation requires metadata, and that means new tools, skills and business processes.
This is mostly a solved problem, but will require learning lessons from other industries
While implementing end-to-end automation requires work, ultimately is means that you spend less time on grunt-work and more time on activities that add value and ultimately help patients!
This Deloitte Insights report, published in Feb 2020, examines the AI technologies in Clinical Trials and is the third in a series, the first is an overview of AI in biopharma and the second is on AI in drug discovery.
AI has the potential to transform key steps of clinical trial design from study preparation to execution towards improving trial success rates, thus lowering the pharma R&D burden.
FDA is already planning for a future in which more than half of all clinical trial data will come from computer simulations.
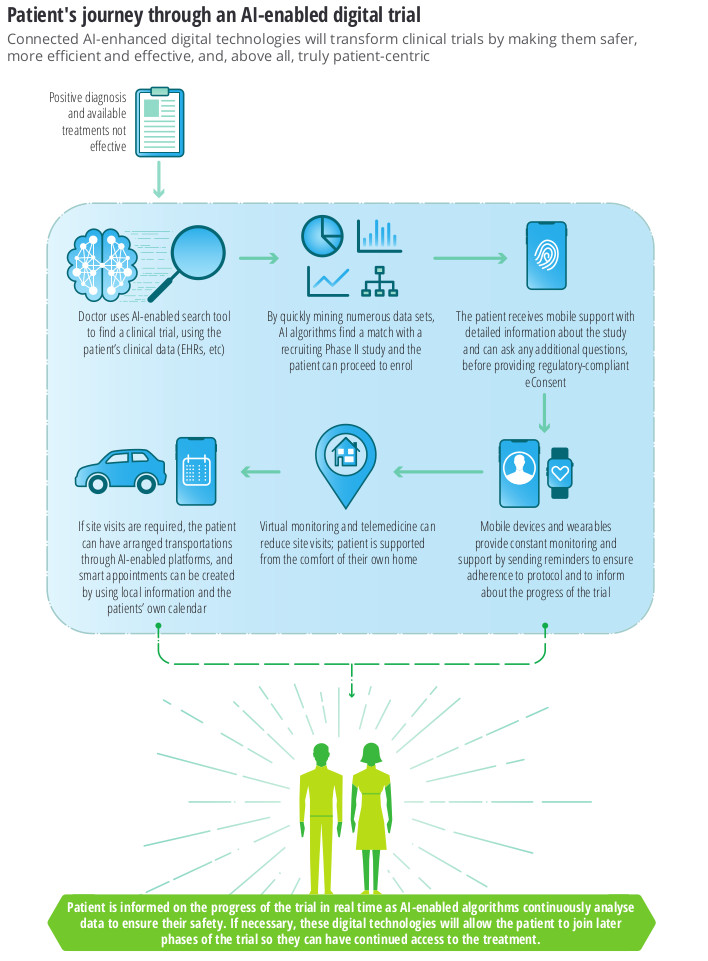
This is a future where phase 1 trials are done in-silico i.e. using a simulation of a human body, and phase II/III trials become remote decentralised clinical trials (RDCT) which use AI-enabled technologies to allow bigger, more diverse and remote populations to participate – as envisioned by the European Innovative Medicines Initiative Trials@Home project, launched in December 2019.
In the coming months, Apple and Google will work to enable a broader Bluetooth-based contact tracing platform by building this functionality into the underlying platforms.
This is an indication of the scale of real-time, real-world data that will be available to enrich regulated clinical trials in the future.
Summary
The benefits of AI for Clinical Trials centre around the optimisation of protocols, patient recruitment and retention
This change will involve enriching clinical data with new sources of real-world data (RWD)
Phase I trials will be run as in-silicocomputer simulations, and phase II/III will become virtual, decentralised clinical trials
Regulatory authorities have already started publishing guidance to industry.
For the next few years, RCT’s are likely to remain the gold standard for validating the efficacy and safety of new compounds in large populations.